
基于基因组数据的癌症药物反应预测:多方法比较与生物学解释
数据处理
✅ 已有数据:
基因表达数据 (
Cell_line_RMA_proc_basalExp.txt)1018个细胞系 × 17737个基因
数值范围:[2.07, 13.96],为log₂转换后的表达值
数据已经过RMA标准化处理
药物反应数据 (
GDSC2_fitted_dose_response_27Oct23.xlsx)969个细胞系:实际参与药物测试的细胞系
286种药物:涵盖多种作用机制的抗癌药物
242,036条测试记录:并非每个细胞系都测试了全部药物
包含IC50(半数抑制浓度)及其对数转换值LN_IC50
细胞系详情 (
Cell_Lines_Details.xlsx)细胞系的组织来源、疾病类型等元信息
药物信息 (
screened_compounds_rel_8.5.csv)621种药物的靶点、通路信息
数据流程图:
原始数据:
├─ 基因表达:1,018个细胞系 × 17,737个基因
├─ 药物反应:969个细胞系 × 286种药物
└─ 细胞系详情:映射表
↓ 预处理
ID映射与对齐:
├─ 映射成功的细胞系(有基因表达)
├─ 细胞系(有药物反应)
└─ 共同细胞系(交集)
↓ 药物选择与特征工程
选择3个药物:
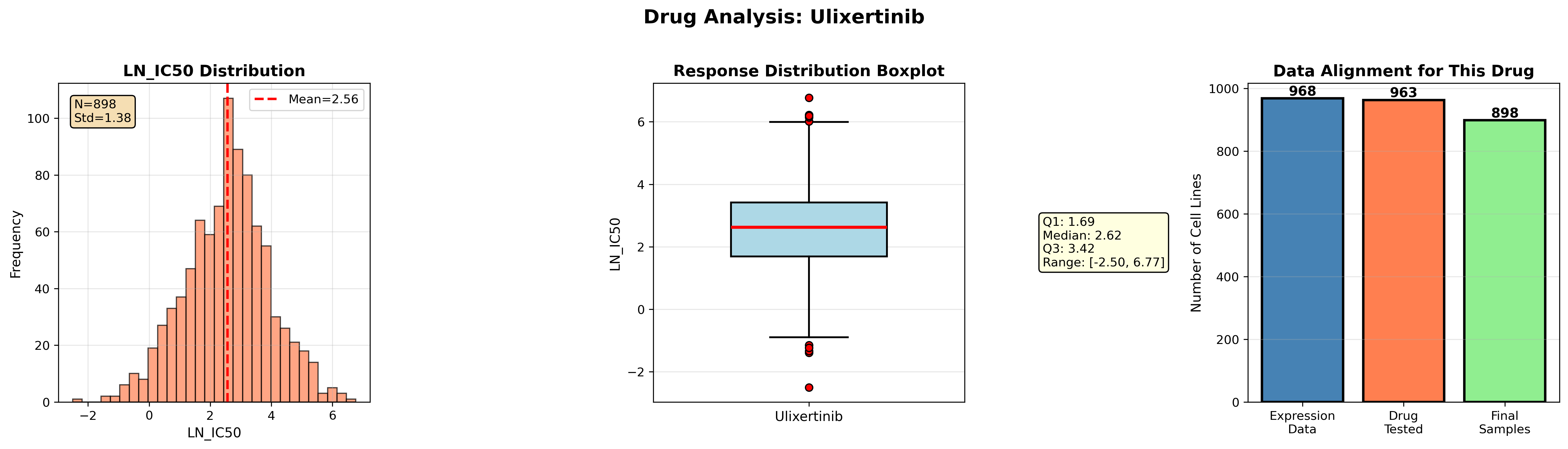
├─ Drug 1: Ulixertinib (898个细胞系,原始963个)
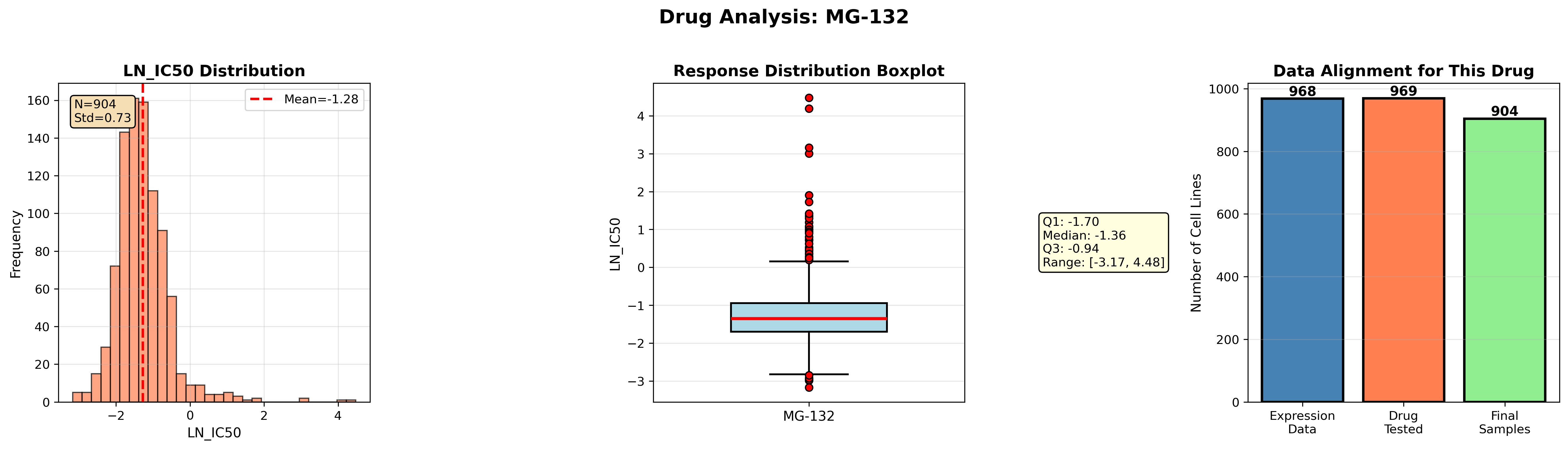
├─ Drug 2: MG-132 (904个细胞系,原始969个)
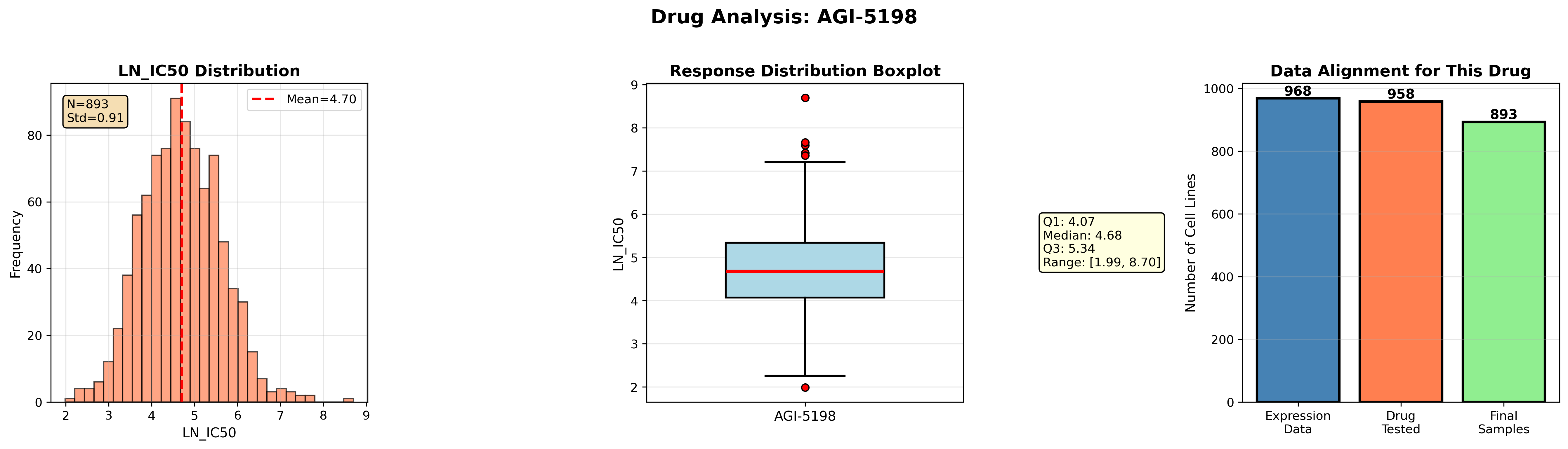
└─ Drug 3: AGI-5198 (893个细胞系,原始958个)
特征选择:
└─ 500个高方差基因(所有药物共用)
↓ 建模数据
Drug 1: X₁ = (898, 500), y₁ = (898, 1)
Drug 2: X₂ = (904, 500), y₂ = (904, 1)
Drug 3: X₃ = (893, 500), y₃ = (893, 1)
其中:
- X = 标准化基因表达矩阵(Z-score)
- y = LN_IC50值(药物敏感性)
↓ 下一步:分别训练3个药物的预测模型数据预处理
1.细胞系ID映射
问题:基因表达数据使用COSMIC数字ID(如DATA.906826),而药物反应数据使用细胞系名称(如CAL-120)。
解决方案:
步骤1: 清理ID格式
'DATA.906826' → '906826'
步骤2: 建立映射字典
使用Cell_Lines_Details.xlsx中的对应关系:
COSMIC ID: 906826 → 细胞系名称: 'CAL-120'
步骤3: 应用映射
将基因表达数据的索引从数字ID转换为细胞系名称映射结果:
原始细胞系数:1,018
映射成功数:968
映射成功率:95.1%
失败原因:50个细胞系在映射表中缺失或ID错误
2.药物选择
筛选标准:
细胞系覆盖数 ≥ 800(保证样本量充足)
LN_IC50标准差在0.5-1.0之间(有区分度但不极端)
不同药物类型(避免机制重复)
最终选择的3个药物:
药物特征分析:
Ulixertinib:Mean=2.56(正值),表明多数细胞系对该药物耐药,适合研究耐药机制
MG-132:Mean=-1.28(负值),多数细胞系敏感,是理想的阳性对照药物
AGI-5198:Mean=4.70(高度耐药),但标准差适中,适合研究特异性敏感亚群
3.数据对齐
对齐逻辑:只保留同时存在于基因表达数据和药物反应数据中的细胞系。
例如对于MG-132药物:
表达数据:968个细胞系(ID映射后)
∩ 交集操作
药物数据:969个细胞系(969个细胞系测试了该药物)
↓
共同细胞系:904个4.特征选择
方法:基于方差的特征筛选
原理:
方差小的基因在不同细胞系间表达几乎一致,无法区分药物敏感性
方差大的基因表达差异显著,更可能与药物反应相关
筛选过程:
原始基因数:17,419(删除318个无名基因后)
↓ 计算方差并排序
选择Top 500个高方差基因
↓
最终特征数:5005.数据标准化
方法:Z-score标准化
公式:
z = (x - μ) / σ
其中:
x = 原始基因表达值
μ = 该基因在所有细胞系中的平均表达
σ = 标准差标准化效果:
标准化前:数据范围广泛,不同基因量纲不一
标准化后:均值≈0,标准差≈1
作用:消除量纲影响,提高模型收敛速度
最终数据集
1.数据集结构
每个药物的数据集包含:
X_features_drugN_*.csv:标准化后的基因表达矩阵
维度:(N个细胞系, 500个基因)
数值:Z-score标准化后的表达值
y_target_drugN_*.csv:药物敏感性指标
维度:(N个细胞系, 1)
数值:LN_IC50(自然对数转换的IC50值)
解读:负值=敏感,正值=耐药
2.生成的数据集清单
processed_data/
├── selected_genes.csv # 500个精选基因列表
├── X_features_drug1_Ulixertinib.csv # 药物1特征矩阵
├── y_target_drug1_Ulixertinib.csv # 药物1目标变量
├── X_features_drug2_MG-132.csv
├── y_target_drug2_MG-132.csv
├── X_features_drug3_AGI-5198.csv
└── y_target_drug3_AGI-5198.csv可视化分析
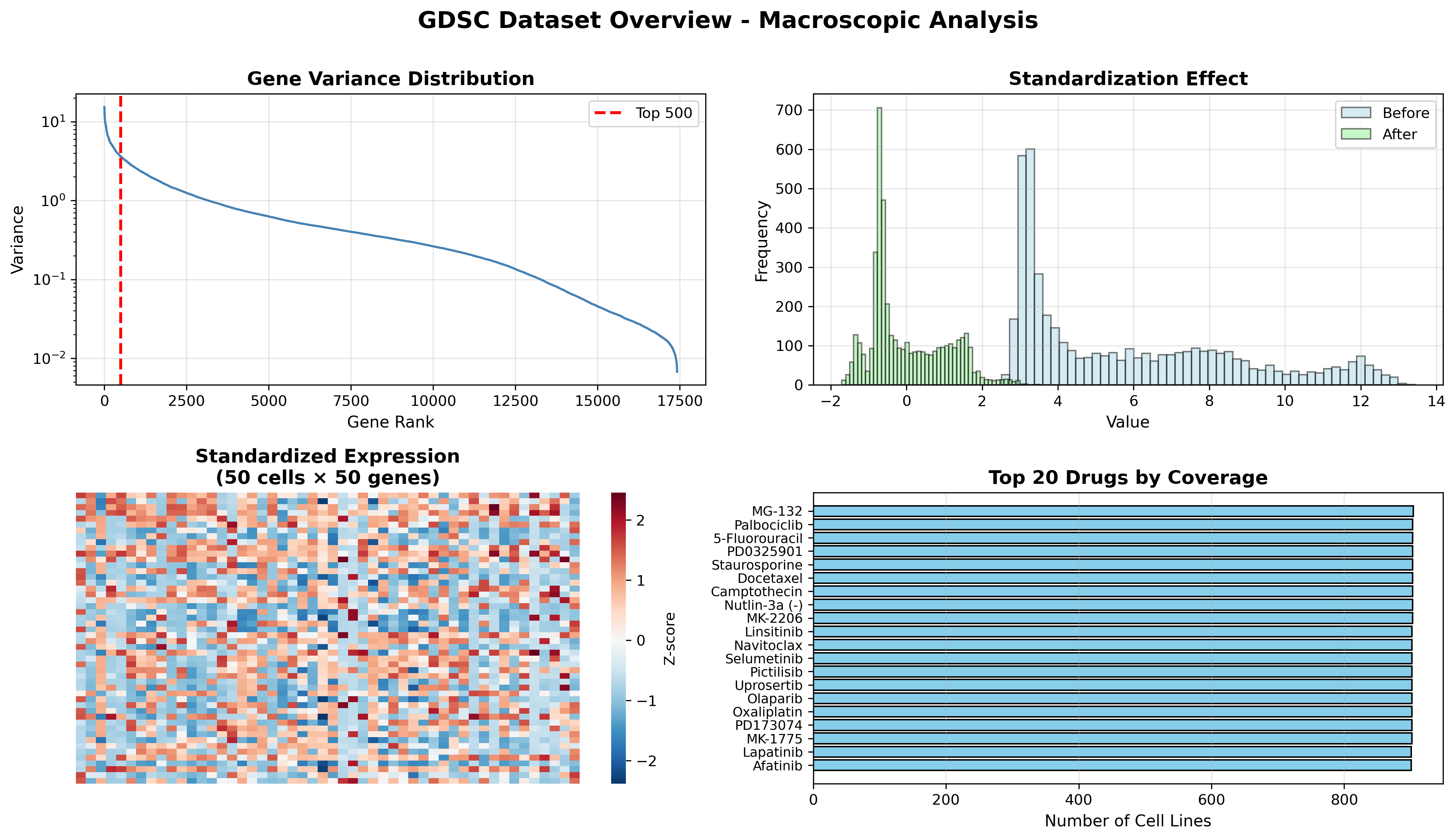
1. 宏观数据特征
4个宏观可视化图:
基因方差分布:展示17,419个基因的方差分布,呈指数衰减,前500个基因包含主要信息
标准化效果:标准化前数据范围[2-14],标准化后集中在均值0、标准差1附近
基因表达热图:50×50采样展示细胞系间表达异质性
Top 20药物覆盖:MG-132等20个药物覆盖900+细胞系
2. 药物特异性分析
为每个药物生成了3类可视化:
1. LN_IC50分布图
Ulixertinib:右偏分布,均值2.56,多数耐药
MG-132:左偏分布,均值-1.28,多数敏感
AGI-5198:右偏分布,均值4.70,高度耐药
2. 响应分布箱线图
展示四分位数、中位数、异常值
用于识别极端敏感/耐药细胞系
3. 数据对齐图
清晰展示从原始数据到最终样本的筛选过程
验证数据完整性
Ulixertinib:
MG-132:
AGI-5198:
Reference
数据来源:
基因表达数据:Pathway Activity Scores - RMA normalised basal expression profiles for all the cell lines
药物反应数据:Drug Download Page - Cancerrxgene - Genomics of Drug Sensitivity in Cancer - GDSC2-dataset
细胞系详情:Drug Download Page - Cancerrxgene - Genomics of Drug Sensitivity in Cancer - Cell-line-annotation
药物信息:Drug Download Page - Cancerrxgene - Genomics of Drug Sensitivity in Cancer - Compounds-annotation
一些自己的疑问记录(无用)
1. 基因表达数据是什么?
准确理解:
1018个细胞系
细胞系 = 实验室培养的永生化细胞株(如HeLa、A549)
每个细胞系代表一种癌症类型
17737个基因 = 每个细胞系测量了17737个基因的表达水平
类比:就像给1018个人做体检,每人测了17737项指标
数据范围 [2.07, 13.96] 的含义:
这是 log2转换后的基因表达值:
原始表达值 → log2(expression + 1) → [2.07, 13.96]
实际含义:
2.07 → 2^2.07 ≈ 4倍表达
13.96 → 2^13.96 ≈ 15,800倍表达为什么取log:
基因表达原始值范围巨大(1到几百万),log压缩尺度
生物学意义:基因表达倍数变化比绝对值更重要
数据更符合正态分布,适合统计分析
2. 药物反应数据详解
242,036条记录 = 969个细胞系 × 286种药物的部分组合
并非每个细胞系都测了全部286种药物,原因:
实验成本高
某些细胞系-药物组合不相关
分批次测试
969 vs 1019的差异:
基因表达数据:1019列(包括1个基因名列 + 1018个细胞系)
↓ 转置删除标题行
1018个细胞系
药物反应数据:969个细胞系(本身就只测了这些)两个数据集来源不同,样本数自然不同。
IC50的含义:
IC50 = 半数抑制浓度(Half Maximal Inhibitory Concentration)
药物浓度 → 0.01μM 0.1μM 1μM(IC50) 10μM 100μM
细胞存活率 → 100% 75% 50% 25% 5%
↑
IC50在这里解读:
IC50 越小 → 细胞越敏感(小剂量就能杀死50%细胞)
IC50 越大 → 细胞越耐药
LN_IC50 = log(IC50):取自然对数,方便建模
3. 细胞系映射过程
是的,用的就是 Cell_Lines_Details.xlsx
映射逻辑:
基因表达数据ID Cell_Lines_Details.xlsx 药物反应数据
'DATA.906826' → COSMIC ID: 906826 → 细胞系名: 'CAL-120'
↓ 映射字典
906826 → 'CAL-120'代码实现:
python
# 步骤1:清理ID
'DATA.906826' → '906826'
# 步骤2:建立映射字典
{
'906826': 'CAL-120',
'687983': 'A549',
...
}
# 步骤3:替换索引
基因表达数据.index = ['CAL-120', 'A549', ...]4. 数据对齐过程
为什么是968个?
步骤1: 1018个细胞系(原始基因表达数据)
步骤2: 映射成功968个(50个细胞系在Cell_Lines_Details中找不到)
失败原因:可能是新增细胞系,或ID错误为什么共同细胞系是904个?
表达数据:968个细胞系(映射后)
∩ 交集操作
药物数据:969个细胞系
↓
共同:904个细胞系
意味着:
- 64个细胞系只有表达数据,没测药物
- 65个细胞系只有药物数据,没测基因表达对齐操作:
python
# 找交集
common_cells = set(['A549', 'HeLa', ...]) & set(['A549', 'MCF7', ...])
↑ 表达数据 ↑ 药物数据
# 只保留共同细胞系
X = 表达数据[common_cells] # 904行
y = 药物数据[common_cells] # 904行5. 为什么只选MG-132一种药物?
这是简化策略,有充分理由:
任务本质:预测"某种药物"的敏感性,不是预测"所有药物"
单药物模型:X(基因) → y(MG-132敏感性)
不是:X(基因) → y(286种药物敏感性)
实际应用:
临床中针对特定药物建模(如"预测患者对紫杉醇的反应")
每种药物的作用机制不同,需要独立模型
MG-132的优势:
覆盖全部904个细胞系(数据最完整)
蛋白酶体抑制剂(重要的抗癌药物类别)
如果要预测多种药物:
可以训练286个独立模型(每个药物一个)
或者用多任务学习(更高级的方法)
你的任务文档中提到"某些药物",意思是选1-3个代表性药物即可。
6. 特征选择基于什么?
基于方差(Variance):
原理:
基因A:所有细胞系表达值都是 [5.0, 5.0, 5.0, ...] → 方差=0 → 无用
基因B:表达值变化大 [2.0, 8.0, 3.0, 9.0, ...] → 方差=10 → 有用逻辑:
方差小 = 细胞系间表达几乎一样 = 无法区分敏感/耐药 = 删除
方差大 = 细胞系间表达差异大 = 可能与药物反应相关 = 保留
计算过程:
python
# 计算每个基因的方差
基因A方差 = 0.01
基因B方差 = 10.5
基因C方差 = 2.3
...
# 排序,取Top 500
选择:[基因B, 基因X, 基因Y, ...](前500个高方差基因)7. Z-score标准化原理
是的,对基因表达数据标准化
Z-score公式:
z = (x - μ) / σ
其中:
x = 原始值
μ = 均值
σ = 标准差举例:
基因A原始值:[2.5, 5.0, 7.5, 10.0]
均值μ = 6.25
标准差σ = 2.87
标准化后:
(2.5-6.25)/2.87 = -1.31
(5.0-6.25)/2.87 = -0.44
(7.5-6.25)/2.87 = +0.44
(10.0-6.25)/2.87 = +1.31作用:
消除量纲影响(不同基因表达范围差异大)
让所有特征在同一尺度(均值0,标准差1)
提高模型收敛速度
8. X_features_scaled.csv内容
完全正确的理解:
904行(细胞系) × 500列(基因)
示例:
RPS4Y1 KRT19 VIM S100P ...
CAL-120 -0.52 1.23 0.84 -1.01 ...
A549 0.88 -0.34 1.56 0.22 ...
HeLa -1.20 0.76 -0.45 1.83 ...
...每个数值是该细胞系中该基因的标准化表达值。
9. y_target.csv内容
一列参数是 LN_IC50(对数化的IC50值)
格式:
LN_IC50
CAL-120 -1.52 ← 这个细胞系对MG-132敏感(负值)
A549 -0.84
HeLa 0.35 ← 这个细胞系对MG-132耐药(正值)
MCF7 -2.10
...解读:
负值越小 → 越敏感(IC50小,低剂量有效)
正值越大 → 越耐药(IC50大,需要高剂量)
这就是机器学习要预测的目标变量(y)。
